

Original paper: Matsuda et al., Mechanoresponsive self-growing hydrogels inspired by muscle training, Science 363, 504-508 (2019)
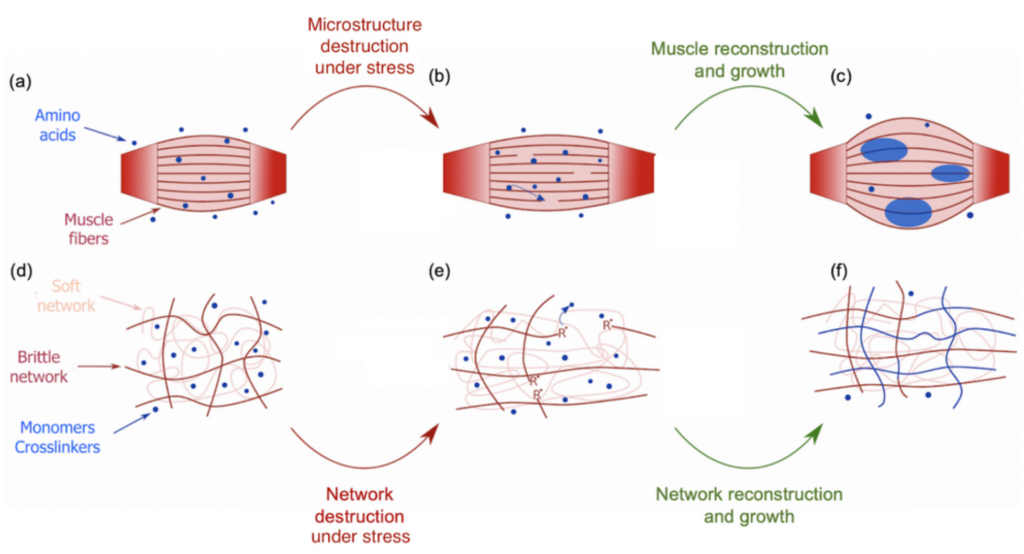
During the COVID-19 lockdown, many of us had the opportunity to workout, for instance by lifting weights to build stronger biceps. During a workout, our muscles undergo some damage at the micrometer scale that triggers an immune system response (Figure 1a). Adequate amino acids, the main constituents of proteins that give muscles their structure, are then carried close to the torn tissues to repair the damage and grow new muscle thanks to the binding between the amino acids and the tissues [1]. This process is a sort of mechanical solicitation where your body creates stronger biological materials with more mass (Figure 1b-c). This seems, however, counter-intuitive when compared to the response of synthetic soft materials under the same mechanical solicitation. Indeed, mechanical stress usually weakens or even damages synthetic materials. For instance, pulling too hard on an elastic band will result in its irreversible failure. Can we then use muscle growth as an inspiration to design materials that would get stronger and bigger under mechanical stress? By mimicking this process, a group of scientists from Hokkaido University developed such a material. This new material belongs to the family of polymer hydrogels. Hydrogels consist of a stretchable 3D network of polymer chains in water connected to each other by molecules called crosslinkers. These crosslinkers control the stiffness of the network. Indeed, a highly connected network will require more stress to be deformed than a loosely connected one.
The difference is that this new material is a double network hydrogel — a hydrogel with two interpenetrated networks — one of them is brittle and rigid due to a high content of crosslinkers, shown as red chains in Figure 1d, while the other one is soft and stretchable due to low content of crosslinkers, shown as pink chains in Figure 1d. This double network is immersed in a solution containing 80-90% water and two types of molecules that are the building blocks of the hydrogel: monomers and crosslinkers. When a tensile stress is applied to the material, the brittle network is the one that “feels” the pressure, leading to breakage of its strands while leaving the soft network undamaged (Figure 1e). During the failure of those polymer chains, the bonds between the carbon atoms break down due to the stress which generates highly reactive chemical species known as radicals [2]. These radicals initiate the formation of new polymer chains by reacting with the building blocks in the solution [3]. As these new chains are also connected by crosslinkers during the reaction, the damaged network is then not only restored but extended and strengthened [4].
The most remarkable result which validates this process is shown in the following movie :
A double network hydrogel is stretched at constant stress three times in a row. The hydrogel is immersed in a solution of monomers and crosslinkers that will be used up for the brittle network reconstruction. After each cycle of stretching, a one-hour reconstruction and growth period allows the material to strengthen. Thanks to that stretching and recovering protocol, the material is able to lift a 200g weight higher and higher as it gets stronger.
The successful emulation of muscle self-strengthening applied to synthetic materials design paves the way for tremendous new bio-inspired materials. They are of especially great interest in soft robotics, where soft materials are exposed to harsh conditions that may lead to damage, like cuts. Rather than undergoing simple regeneration, the material would respond to damage by strengthening to better resist its environment.