
Original paper: The fractal globule as a model of chromatin architecture in the cell
The entirety of our genetic information is encoded in our DNA. In our cells, it wraps together with proteins to form a flexible fiber about 2 metres long known as chromatin. Despite its length, each cell in our body keeps a copy of our chromatin in its nucleus, which is only about 10 microns across. For scale, if the nucleus was the size of a basketball, its chromatin would be about 90 miles long. How can it all fit in there? To make matters worse, the cell needs chromatin to be easily accessible for reading and copying, so it can’t be all tangled up. It’s not surprising then that scientists have been puzzled as to how this packing problem can be reliably solved in every cell. The solution is to pack the chromatin in a specific way, and research suggests that this may be in the form of a “fractal globule”.
An equilibrium globule is the state that a polymer (a long repetitive molecular sequence, like chromatin) takes when it is left for a long time in a liquid that doesn’t dissolve it well. In such a liquid, the polymer is more attracted to itself than the molecules around it, so it collapses into a globule to minimize the amount of contact between itself and its surroundings. The resulting object is much denser than typical polymers in good solvents and is dense enough to fit inside a nucleus. However, like stuffing headphone cables into your pocket, it develops many knots and its different regions mix with one another.
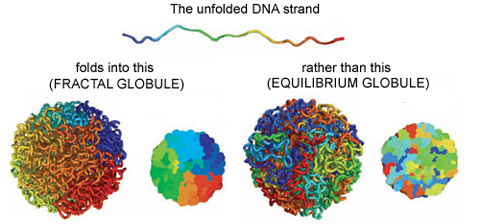
On the other hand, if you change the polymer’s environment fast enough that it doesn’t have the time to fully equilibrate, then every piece of the polymer will locally collapse into its own globule. In other words, the polymer forms a globule made of smaller globules and is called a fractal globule. Fractals are objects which look the same at all scales, like the edge of a cloud or the coastline of England. If you zoom in or out on either of these objects, they look more or less the same. This isn’t an “equilibrium” state, meaning it will slowly fall out of this configuration. However, until the whole polymer equilibrates (which takes a long time), the chain has many desirable properties.
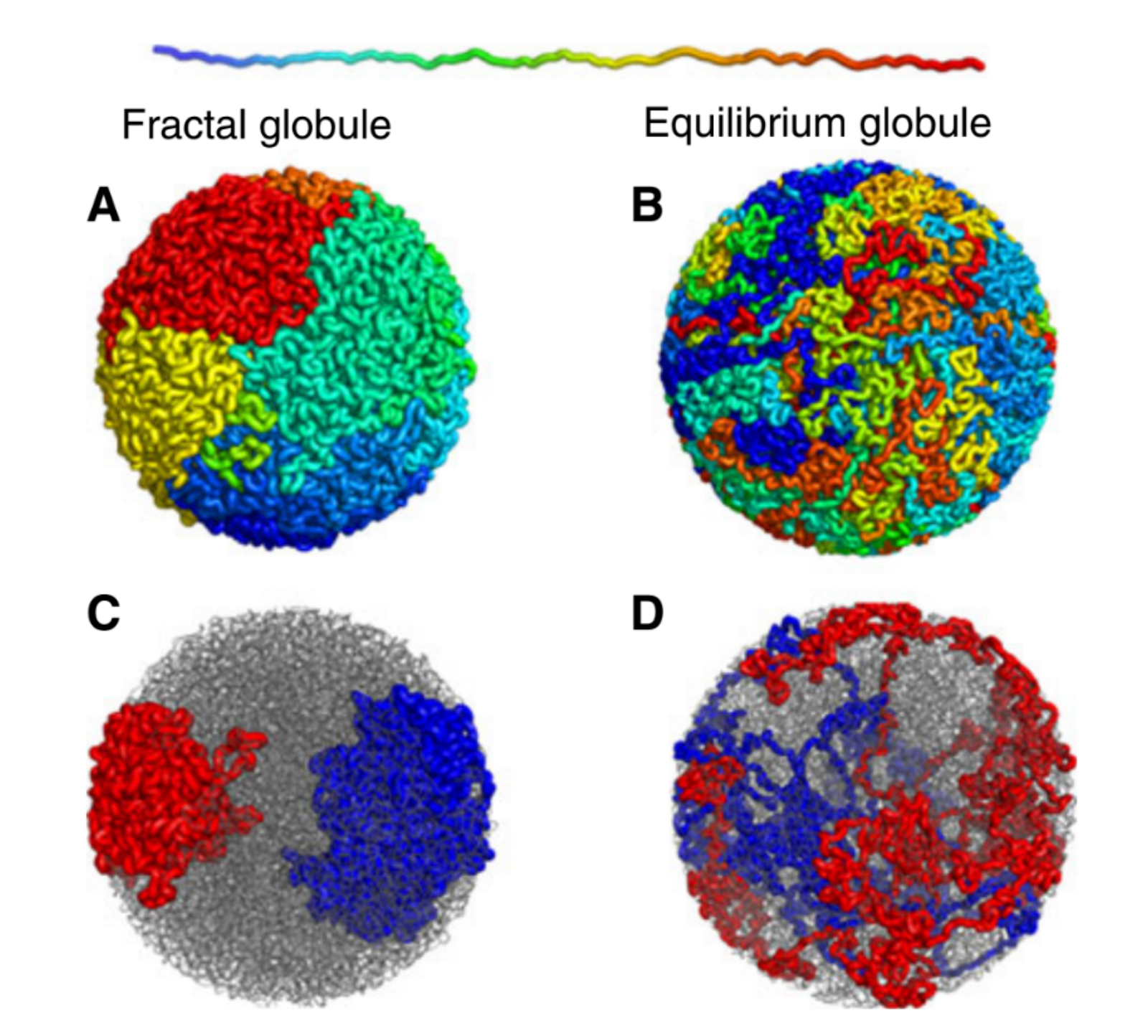
Figure 1. Simulated examples of fractal (A,C) and equilibrium globules (B,D), showing compartmentalization of different portions of the polymer. The chain color goes from red to blue as shown above. Compartmentalization means that parts of the chromatin stay near other parts with the same color (adapted from paper [1]).
We are interested in these globule states because they are dense enough that a globule of chromatin can fit inside of a cell nucleus. But it’s not enough to simply fit inside; the cell needs chromatin to avoid forming knots, since getting tangled would prevent the cell from properly reading its own DNA. Live cells also keep their chromatin nicely compartmentalized, that is, different regions along the genome stay spatially separated from one another. Unlike equilibrium globules, fractal globules have few knots and are also compartmentalized! To get a better picture for what this means, Leonid Mirny performed simulations of the different types of globules. Figure 1 shows the results of these simulations, highlighting how different the two states look in terms of knotting and separation of regions of the polymer.
So it seems that the fractal globule state has all the properties we need for a good model of chromatin! But, as scientists, we know that no matter how well a theory fits the characteristics we want it to have, we need experimental evidence before believing anything. In the case of this fractal globule model for genome organization, evidence has come in the form of “contact probability maps”. These are collected from large populations of cells whose DNA is cut, spliced, and read in such a way that allows for a measurement of the probability that any two sites on the chromatin are touching at any given time. Among other things, these maps give us information about how chromatin is packed. So the question becomes, “what does the fractal globule model predict a contact probability map to look like?”
The fractal globule model doesn’t make exact predictions about where one will find specific segments of chromatin, but it does predict a contact probability as a function of distance between two sites, s. Specifically, the model predicts that the contact probability between two sites scales like 1/s. Meaning, if I look at sites that are twice as far apart along the polymer, then they are half as likely to be touching. This 1/s scaling is what was observed on intermediate scales (about 100,000 to 6 million base pairs) by looking at contact probability maps averaged over a whole population of cells.
We still don’t know how the cell maintains and tunes this fractal globule state, and we still have not developed a dynamic version of this picture, which is necessary since it is well-established that the chromatin in our cells is far from static. But this study gives us a new picture of how chromatin is organized inside cells. It isn’t randomly configured like headphone cables in your pocket or a ball of yarn. Rather it is folded onto itself in a self-similar way. This model is attractively simple, requires little fine-tuning, all while producing a long-lived state with segregated territories and easily accessible genes.