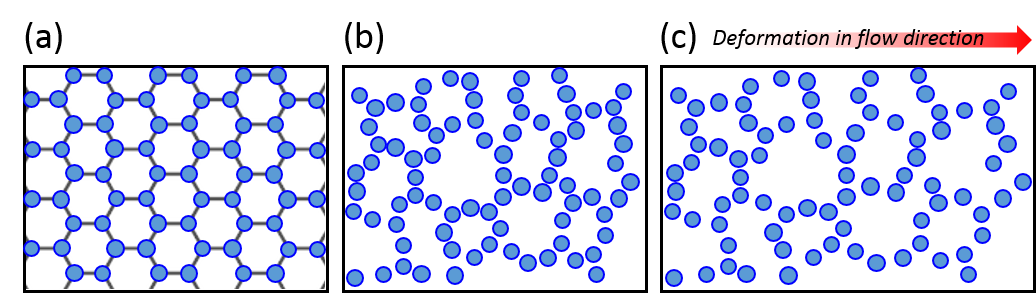


Original paper: Magnetically Addressable Shape-Memory and Stiffening in a Composite Elastomer
Video 1. Heat-shrink tubing demonstration.
Have you ever used heat-shrink tubing at home to seal an exposed wire? As shown in Video 1, you would place the tubing around your wire, apply heat, and voilà! The tubing shrinks and tightly wraps itself onto the exposed wire, and you don’t have to worry about an electric shock anymore. This type of material that changes its shape upon increased temperature is called a shape-memory polymer. Since its commercial development in 1962, scientists have found this type of material so useful that its popularity rose, especially in the biomedical and aerospace fields. However, it comes with a few drawbacks: applying the desired temperature uniformly can be tricky and the shape change induced by the heat can be quite slow. In addition, changing the temperature isn’t ideal for biological applications where the environment surrounding the material is sensitive to heat, such as in tissues and living cells. In today’s post, I’ll introduce you to a different type of shape-memory material that “remembers” its temporary shape when subjected to a magnetic field, instead of heat.
Video 2. Demonstration of the material developed by the authors.
Paolo Testa and coworkers from the Paul Scherrer Institute and ETH Zurich developed a shape-memory polymer composite that can preserve a new shape when a magnetic field is present. As you can see in Video 2, the initially flexible material sitting on the center stage is manually twisted with a tweezer and held by force. When the magnetic ring is raised around the stage and held up so that the material can “feel” the magnetic field, the material “freezes” in its twisted shape, even when the tweezer is removed. After a certain time, the magnetic ring is removed and the material regains its original shape within one second, dramatically faster than the time it took for a heat-shrink tubing in Video 1 to change its shape. The material—at least a part of it—that looks like black rubber is a flexible polymer that is the main ingredient of the Silly Putty, called poly(dimethylsiloxane) (PDMS). However, PDMS is normally transparent in color, and PDMS itself isn’t a shape-memory polymer. So what makes it black and capable of holding the twist when the magnetic field is present?
The answer is in iron particles. However, iron particles alone cannot perform well enough. Shape-memory materials that were previously developed had iron particles directly embedded in the polymer but didn’t have such a high sensitivity to magnetic fields. What makes the material in this paper so unique is the liquid surrounding the particles. The iron particles are dispersed in a water and glycerol mixture, making the fluid six times more viscous and stiff when subjected to the magnetic field. This type of fluid, called a magneto-rheological fluid, is then injected into the PDMS polymer, making the material sensitive to the presence of a magnetic field.
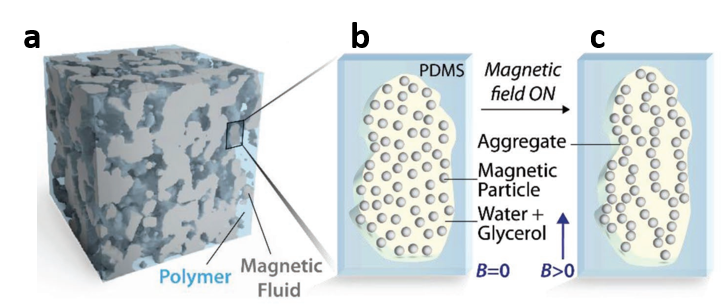
Figure 1 shows the 3D structure of the developed material, as well as a 2D schematic of a magneto-rheological fluid droplet with and without the magnetic field. When the fluid is injected into the polymer, the polymer encases the fluid and a composite is formed, which is shown in Figure 1a. In the absence of the magnetic field, shown in Figure 1b, the iron particles are dispersed and mobile inside the fluid droplet. However, when the magnetic field is turned on, shown in Figure 1c, the particles reorganize and align along the direction of the magnetic field, and, in turn, the droplet stiffens. The alignment of the iron particles and the resulting stiffening of the fluid, induced by the presence of a magnetic field, are the reasons why the polymer composite can hold its new shape (as in Video 2). When the magnetic field is removed, the particles regain their mobility and the fluid droplets soften, which lets the polymer return to its original shape.
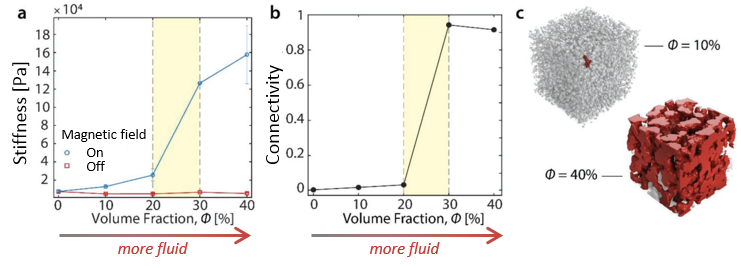
By controlling the ratio between the magneto-rheological fluid and PDMS (?), the authors tried to understand the relationship between the stiffness and structure of the material and the said ratio. When the percentage of the fluid increases, as shown in Figure 2a, the overall stiffness of the material measured in the presence of the magnetic field increases as well. This increase is enhanced when fluid occupies more than 20% in volume, as highlighted in yellow in Figure 2a. Compared to a factor of two increase going from 10% to 20%, the stiffness increases by a factor of 13 going from 10% to 30%.
By measuring the connectivity [1] between the fluid droplets using an X-ray scan [2], the authors discovered that the stiffness increase is related to the network connection between the fluid droplets when the magnetic field is applied (Figure 2b). This network connection is visualized in Figure 2c; the fluid, shown in red, is isolated when the fluid composes only 10%. However, when increased to 40%, the dispersed fluid pockets become connected to one another, enhancing the stiffness of the overall material under the magnetic field.
As mentioned above, the addition of the fluid was the key to the material’s drastic functional improvement. Since the fluid provides freedom for the iron particles to move around and align under the magnetic field, the stiffening process becomes more dramatic compared to having the particles alone inside the polymer. Also, the fluid acts as a buffer, lessening any damages caused to the polymer by the motion of the hard particles. The authors hope that their research will open up an even wider range of applications using this shape-memory polymer, such as a magnetic-controlled micromachine that can deliver drugs to targeted areas inside our body. Unlike the temperature-controlled shape-memory material introduced in the beginning, the stiffening of this new magnetically controlled shape-memory material is reversible. This might offer a greater potential for new applications which might require several cycles of deformation. Maybe in the near future, we’ll use items made out of magnetically controlled shape-memory polymers in our daily lives.
[1] The connectivity is defined as the volume of the biggest droplet over the total volume of the magneto-rheological fluid in the polymer composite.^
[2] The authors used X-ray tomography, a method where a 3D image is constructed using 2D X-ray images.^